
I. A SYSTEM UNDER STRAIN — OR ONE BEING WORKED AROUND?
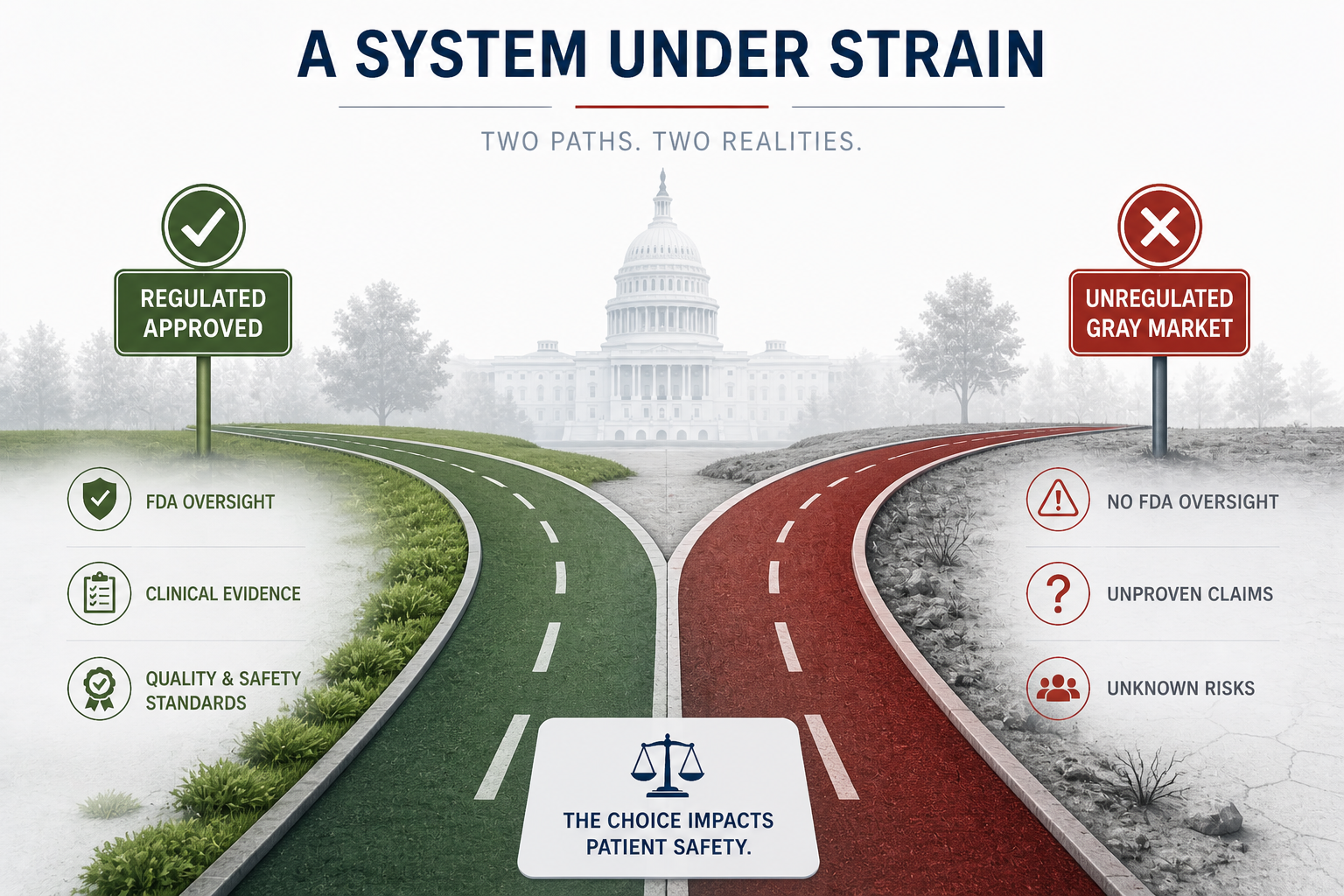
Across the United States, a multi-billion-dollar regenerative medicine market has quietly evolved into something far less controlled than many patients realize. What is marketed as “cutting-edge stem cell therapy” often exists in a regulatory gray zone—where federal law is clear, but enforcement is inconsistent, and state-level legislation increasingly muddies the waters.
At the center of this issue is the U.S. Food and Drug Administration (FDA), which has repeatedly drawn a hard line:
-
Only a narrow class of stem cell products—hematopoietic (blood-forming) cells—are FDA-approved in the U.S.
-
The vast majority of products marketed as “stem cells,” “exosomes,” or “birth tissue biologics” are unapproved drugs or unverified biologics
And yet they are being sold, injected, and infused into patients every day with no quality control or oversight.
II. THE ILLEGALITY OF IV STEM CELL AND EXOSOME TREATMENTS
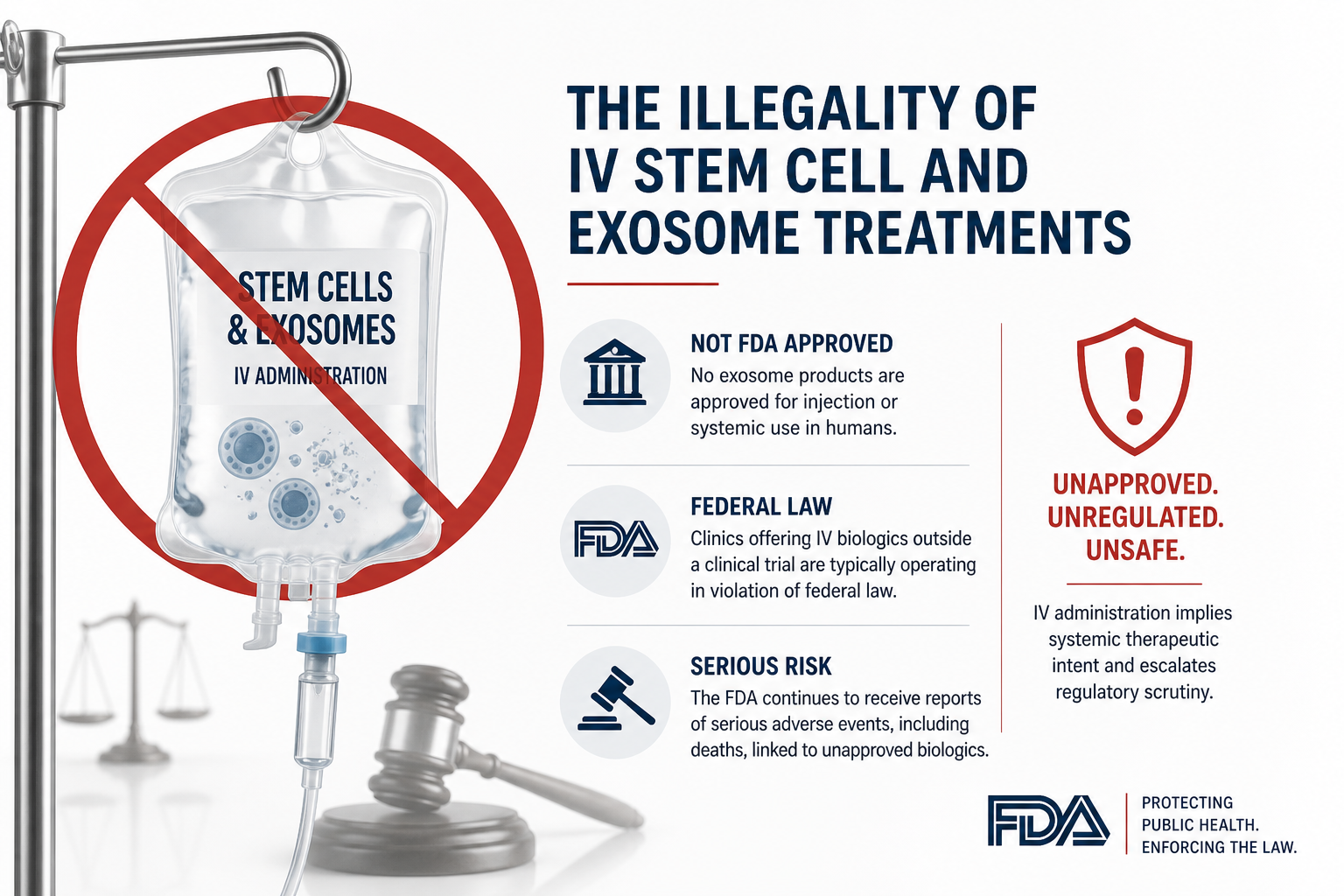
The most significant, and often misunderstood, line in U.S. law comes down to how these products are used.
Under FDA rules:
-
Products that are “more than minimally manipulated” or used for non-homologous purposes must undergo full drug approval pathways. This has not happened in the U.S. and remain entirely illegal and without oversight.
-
IV (intravenous) administration automatically escalates regulatory scrutiny, as it implies systemic therapeutic intent.
This is where the industry fractures.
The FDA has made its position explicit:
-
No exosome products are approved for injection or systemic use in humans
-
Clinics offering IV biologics outside a clinical trial are typically operating in violation of federal law
Even more concerning:
-
The FDA continues to receive reports of serious adverse events, including deaths, linked to unapproved biologics as they are being created or shipped in a black market and avoid all saftey controls.
This is not theoretical risk, it is documented reality and why we are sharing here.
III. THE PLATINUM BIOLOGICS CASE — A WINDOW INTO THE INDUSTRY

At the center of recent enforcement is Platinum Biologics—a company that illustrates how far the gap between regulation and practice has stretched.
The FDA formally concluded that Platinum Biologics:
-
Sold unapproved new drugs in violation of the Federal Food, Drug, and Cosmetic Act
-
Distributed unlicensed biological products without a Biologics License Application (BLA)
-
Marketed products for treatment of diseases including autoimmune conditions, pain, and tissue repair—triggering drug classification
More critically:
-
Their umbilical cord-derived products were deemed “more than minimally manipulated”, disqualifying them from lighter regulatory pathways
-
The transformation into injectable or flowable products altered the original biological structure, a key regulatory violation
In plain terms:
They were manufacturing and selling biologic drugs without approval. And doing so in lyophilized form. What is lyophilized you may ask?
IV. POWDERIZED (LYOPHILIZED) PRODUCTS — SCIENCE OR MISREPRESENTATION?

One of the most controversial practices involves lyophilization—freeze-drying biologic material into a powder.
Companies like Platinum Biologics have marketed these products as:
-
“Stem cells”
-
“Exosomes”
-
“Regenerative biologics”
But here is the scientific reality:
-
Living mesenchymal stem cells (MSCs) cannot survive lyophilization without losing viability
-
What remains is cellular debris, proteins, or signaling remnants—not living stem cells
Yet these products are frequently:
-
Reconstituted
-
Injected
-
Marketed as “live cell therapy”
This is not a minor discrepancy—it is a fundamental misrepresentation of biological function.
V. THE SUPPLY CHAIN: TISSUE SOURCING AND TRACEABILITY RISKS
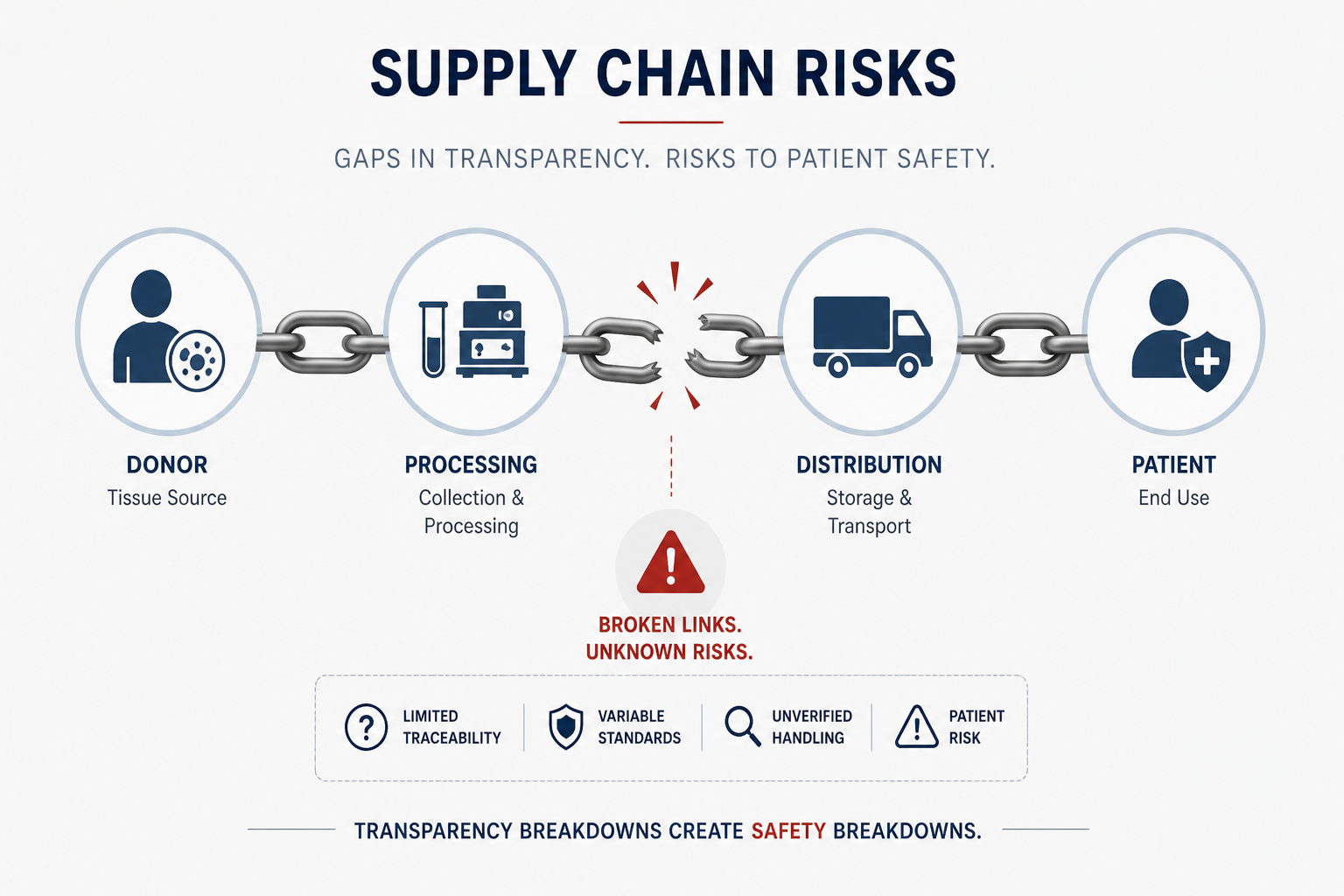
While companies like Platinum Biologics distribute finished products, upstream sourcing often involves third-party tissue providers such as TriLife (who are often referenced in industry discussions).
The concerns here are systemic:
-
Chain-of-custody transparency is often limited
-
Donor screening, processing, and handling standards vary widely
-
Products may be:
-
pooled
-
heavily processed
-
altered beyond original biological intent
-
Under FDA rules, once tissue is:
-
manipulated
-
repurposed
-
or marketed for new therapeutic uses
…it becomes a regulated drug product, not a simple tissue transplant.
And yet, much of the market still operates as if it does not.
VII. THE BROADER PATTERN: WARNINGS, NOT ABSENCE OF LAW

The narrative is often framed incorrectly:
“There are no rules.”
That is not true.
The reality is more concerning:
There are rules—but they are being bypassed, misunderstood, or selectively ignored.
The FDA has:
-
Issued multiple warning letters to companies selling birth tissue and exosome products
-
Publicly warned patients against unapproved regenerative therapies
-
Documented serious harm linked to these interventions
And still—the market continues to grow.
VIII. THE CONSEQUENCE: PATIENT RISK DISGUISED AS INNOVATION

Patients are often told:
-
“These are stem cells”
-
“They’re safe”
-
“They’re cutting-edge”
But in many cases, what they are receiving is:
-
Unapproved biologic material
-
With uncertain composition
-
Delivered in ways explicitly flagged by regulators as unsafe or illegal
This is not innovation.
It is regulatory arbitrage.
IX. WHERE TRUE OVERSIGHT DIFFERENTIATES — THE AURAGENS MODEL

Against this backdrop, the distinction is not marketing—it is infrastructure.
What separates high-integrity programs is not the claim of “stem cells,” but the system surrounding them:
1. Full QA/QC and Laboratory Rigor
-
Defined manufacturing standards
-
Batch traceability
-
Controlled processing environments
2. Regulatory Alignment (Not Avoidance)
-
Built around international frameworks that allow advanced therapies within structured oversight
-
AABB Accreditation (FDA deemed status organization)
3. Product Integrity
-
Viable, characterized cell populations
-
Not lyophilized or degraded substitutes
-
ISO Lab Certification
-
cGMP compliant
4. Clinical Intent and Execution
-
Physician-led protocols
-
Indication-driven treatment—not broad, catch-all marketing claims
5. Transparency
-
Clear differentiation between:
-
research
-
approved therapies
-
investigational use
-
X. FINAL THOUGHT: THE INDUSTRY AT A CROSSROADS

The U.S. regenerative medicine space is at an inflection point.
On one side:
-
rapid innovation
-
expanding demand
-
real therapeutic potential
On the other:
-
misrepresentation
-
regulatory gaps
-
patient risk
The FDA has already drawn the line.
The question is not where the law stands—
but who is choosing to operate within it.

Auragens exists to be the bridge. The safest way to access the most advanced regenerative therapies in the world. With its own biologics created on-site and going through rigorous third-party testing prior to reaching a patient. Not only did Auragens create the best quality biologic, they did so with full regulatory oversight and deliver it in a luxurious, five star clinical setting.
About Auragens










{kind=link}